CAR-T Cell Transplant Multiple Myeloma,FDA ODAC Votes in Favor of Two CAR-T Therapies for Early Treatment of Multiple Myeloma
CAR-T Cell Transplant Multiple Myeloma,FDA ODAC Votes in Favor of Two CAR-T Therapies for Early Treatment of Multiple Myeloma
BCMA CAR-T Discussion
At 8:30 pm Beijing time on March 15, 2024, the FDA’s Oncologic Drugs Advisory Committee (ODAC) officially convened, with the meeting lasting over 9 hours. The focus of this meeting was to discuss the expanded indication applications of two BCMA CAR-T cell therapy products in the treatment of multiple myeloma (MM).
The first product was Abecma from BMS/2seventy bio, which was already approved by the FDA in 2021 for the treatment of relapsed or refractory multiple myeloma (R/R MM) patients after five prior lines of therapy (including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 antibody).
In April last year, based on the results of the KarMMa-3 study, Abecma’s application for the indication of third-line treatment of R/R MM was submitted to the FDA, with an original target action date of December 16, 2023, which was later delayed.
The second product was Carvykti from Legend Biotech/Johnson & Johnson, which was approved by the FDA in February 2022 for the treatment of R/R MM patients after five prior lines of therapy (including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 antibody).
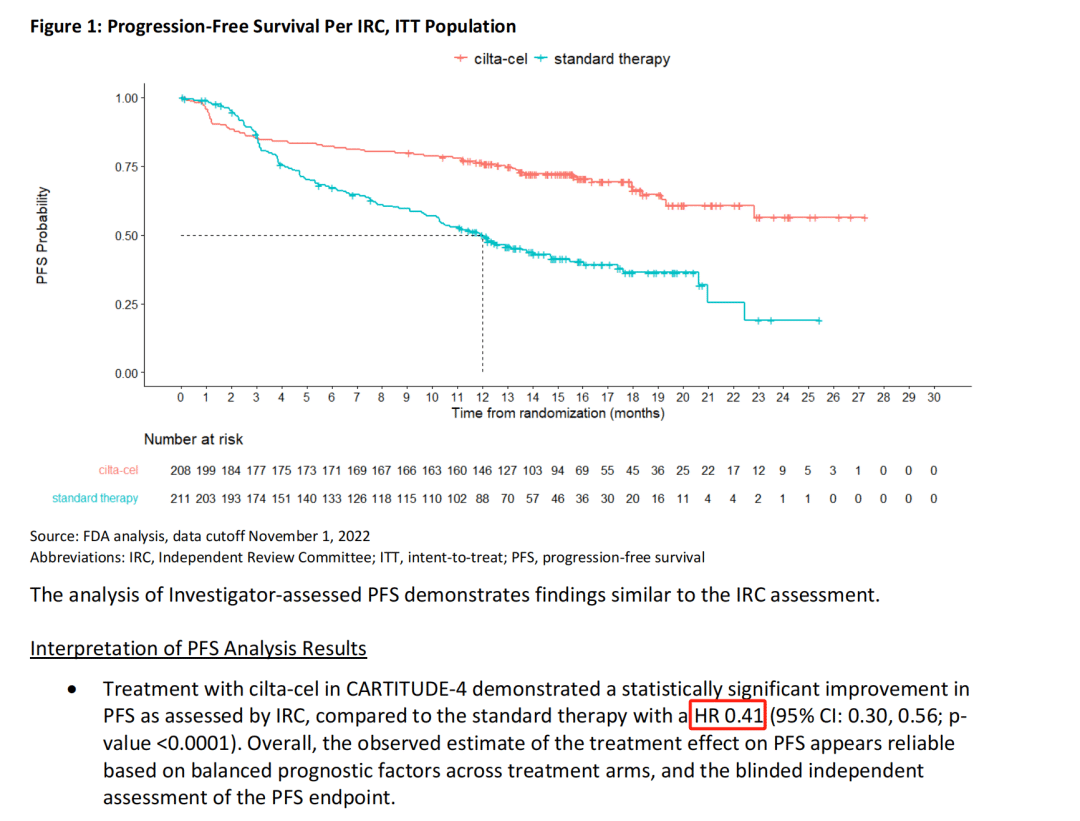
In June last year, based on the results of the CARTITUDE-4 study, the application for Carvykti’s indication for second-line treatment of R/R MM was submitted to the FDA, with an expected action date of April 5, 2024.
During the review process, the FDA found that although both products demonstrated promising long-term benefits, patients in the CAR-T treatment groups in both Phase III clinical trials showed an increased risk of early death compared to the control groups.
For late-stage MM patients who have undergone multiple lines of treatment and are unresponsive to existing standard therapies, the available treatment options have very limited efficacy. Compared to the long-term substantial benefits, the early risks associated with CAR-T therapy are acceptable.
However, when it comes to second-line or third-line treatment, the choice becomes more challenging. Particularly at the beginning of this year, the “CAR-T therapy-induced secondary T-cell malignancy” issue raised FDA’s attention, and although the academic community unanimously agreed that it was not a significant concern, the FDA still added a black box warning to relevant products, making the agency more cautious about approving CAR-T therapies.
During the meeting, 11 experts voted on the issue of “whether to support the relevant CAR-T indication applications based on the known risks and benefits,” and the results were 11:0 and 8:3 in favor of the indication applications for Carvykti and Abecma, respectively.
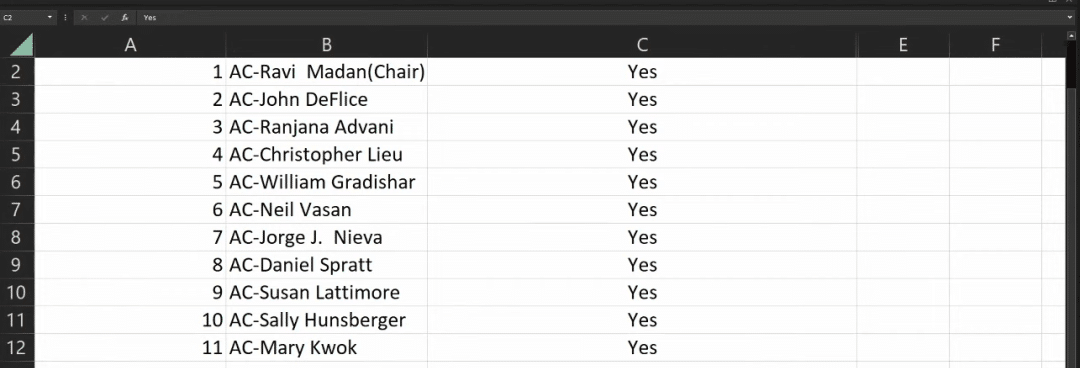
Carvykti voting results
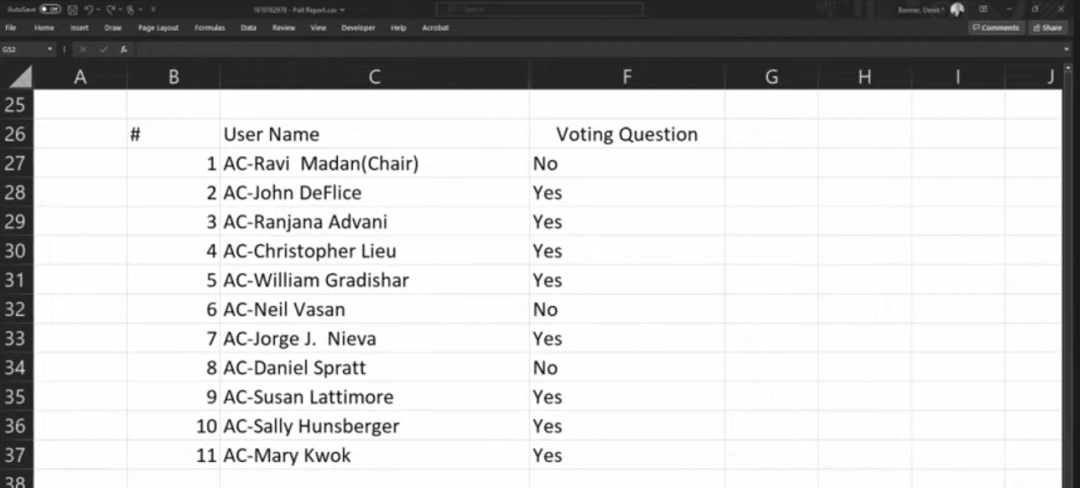
Abecma voting results
Early Death Risk VS Long-term Excessive Benefit
Both CAR-T therapies faced the same issue: the risk of excessive early mortality. This is related to the treatment pathway and preparation cycle of CAR-T therapy.
Both CAR-T therapies are autologous cell therapies, which involve the customization of cell therapy for each patient, including the collection, modification, and expansion of the patient’s own T cells. This process often takes several weeks, creating the so-called “treatment window.” Patients may experience irreversible disease progression while waiting for the CAR-T cell product to be ready, thereby increasing the possibility of early death.
Even though patients receive “bridging therapy” during this period, it is difficult to prevent disease over-progression, as the purpose of bridging therapy is to stabilize and control the patient’s condition, and the treatment regimen is designed to avoid interference with the subsequent CAR-T cell therapy activity, which limits its effectiveness.
Furthermore, prior to CAR-T therapy administration, lymphodepletion chemotherapy is required, which can lead to a significant decrease in the patient’s immune function in the short term, increasing the risk of infection and complications.
For Carvykti, after 8 weeks of randomization, the risk of disease progression or death in the trial group was reduced by 74%, which is a very impressive result.
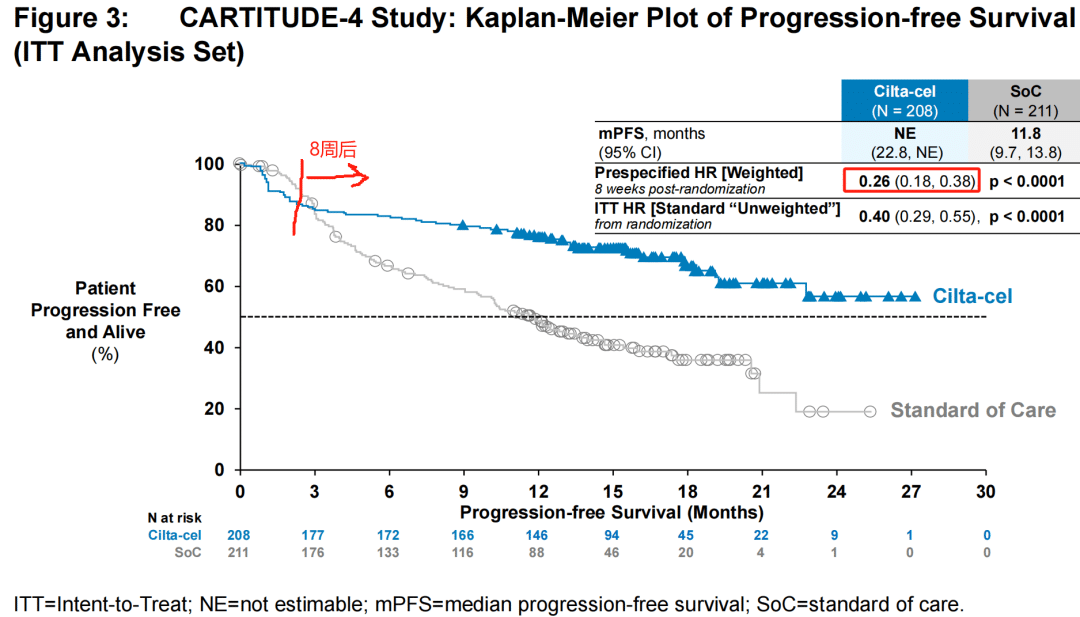
Even when considering the bridging therapy period and including patients who experienced disease over-progression, the risk of disease progression or death in the trial group was still reduced by 59%.
Subgroup analysis showed that regardless of the patients’ baseline condition, Carvykti significantly reduced the risk of death.

The challenge lies in the fact that this “early” period is quite long. It was not until the 11th month that the survival curves of the trial group and the control group began to cross. Generally speaking, even accounting for the time required for CAR-T preparation, bridging therapy, and lymphodepletion, the time from patient enrollment to CAR-T infusion treatment is unlikely to exceed 2 months.
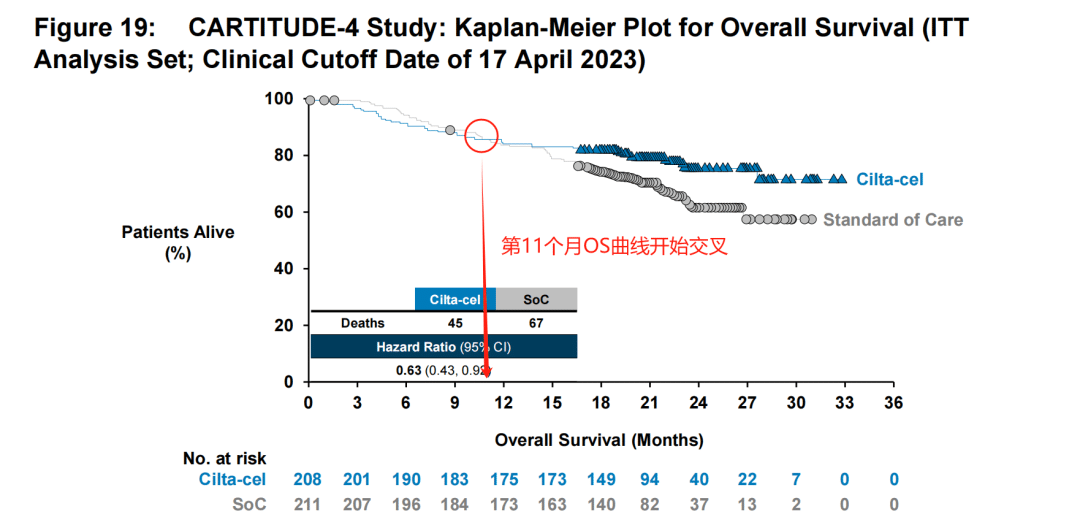
This means that patients still need to endure 9 months before they can experience a benefit over the standard of care (SoC). Does this imply that the adverse effects of CAR-T cells are too severe? This is the concern of the FDA.
The explanation for this is as follows: First, based on the stage analysis, the survival benefit brought by CAR-T therapy has already begun from the 6th month onwards.
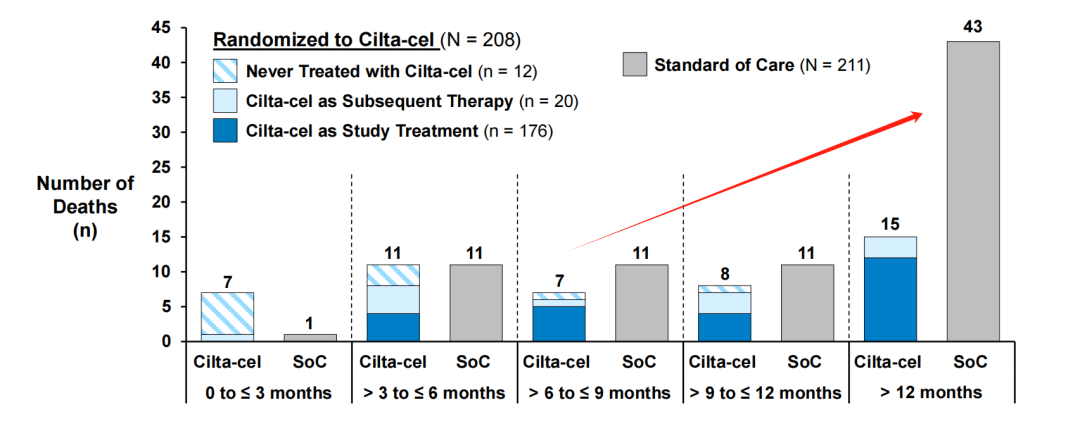
Secondly, although the safety analysis showed that 11% of patients receiving Carvykti died due to treatment-related adverse events, higher than the 8% in the control group, 3.7% of the deaths in the trial group were caused by COVID-19, while the proportion in the control group was 0.5%. In other words, drug side effects are not necessarily the direct cause of treatment-related adverse events.
Looking at the overall survival data, at the second interim analysis, the median OS in the trial group was not reached, while the estimated median OS in the standard treatment group was 26.7 months (HR=0.78, 95% CI: 0.51-1.20).
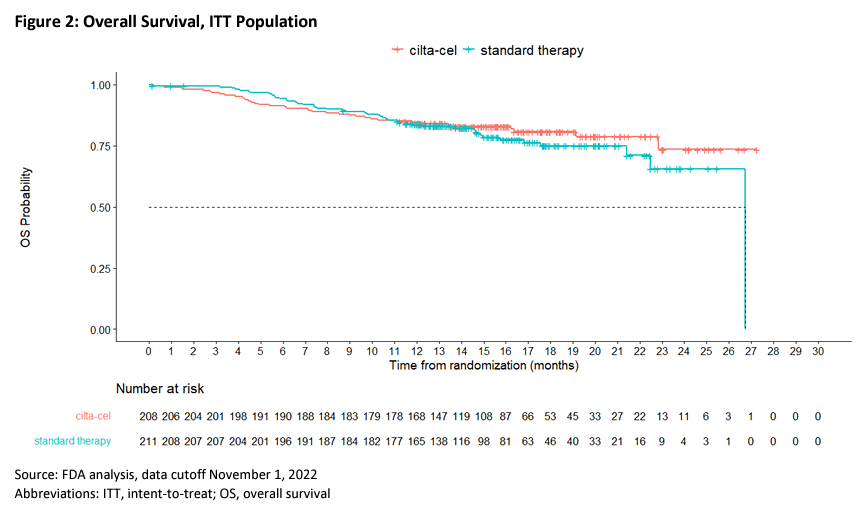
The experts present accepted Legend/Johnson & Johnson’s explanation and did not believe that the early risk should be attributed to the treatment itself, and the long-term PFS was convincing. Compared to the tremendous benefits of treatment, the “very real but small risk” of early death is acceptable.
Based on the briefing materials provided by the FDA before the meeting and the final voting results, it appears that the experts were more concerned about Abecma compared to Carvykti.
In the KarMMa-3 study, although ide-cel demonstrated significant advantages in PFS and ORR, the researchers used a crossover study design, allowing patients in the standard treatment group to cross over and receive Abecma after disease progression confirmation, which complicated the interpretation of the OS results.
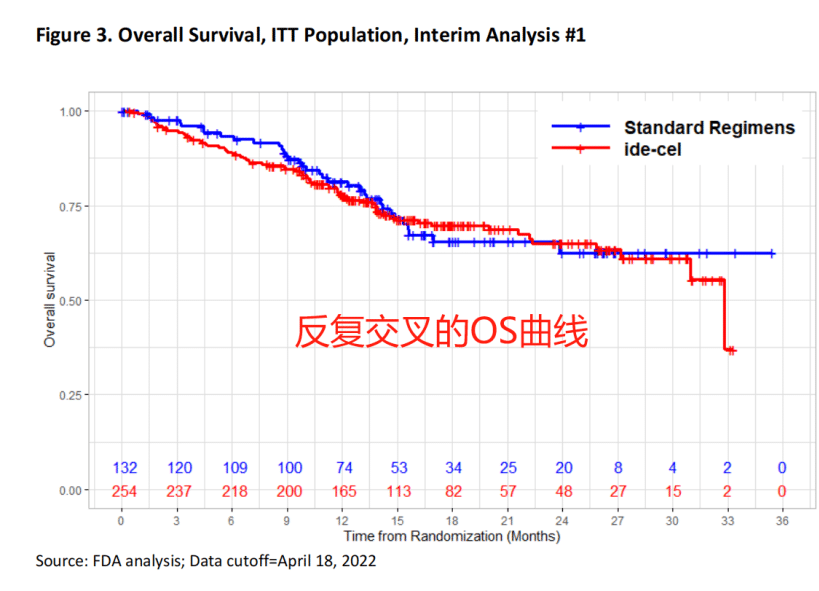
Furthermore, the FDA stated that the subgroup analysis data provided by the researchers did not “adequately describe” the characteristics of the heterogeneous patient population.
The experts also expressed hesitation about Abecma, stating that the bridging therapy seemed problematic, and the survival curve was somewhat “unexplainable.” However, considering the long-term PFS benefit, Abecma still passed the ODAC meeting.
Supplementary Information
About the ODAC Meeting
The Oncologic Drugs Advisory Committee (ODAC) is an independent expert advisory team under the U.S. Food and Drug Administration (FDA), specifically responsible for providing scientific and medical advice to the FDA on the research, development, approval, and regulation of anticancer drugs. The main functions of the ODAC meeting include: reviewing and evaluating data, providing decision-making recommendations, guiding regulatory policies, promoting public participation and transparency, resolving disputes and uncertainties.
However, the voting decisions of the ODAC meeting and other expert advisory committees do not represent the FDA’s final approval opinion. It has happened that the FDA’s review results have “contradicted” the expert advisory committee’s decisions.
About the ODAC Meeting Briefing Materials
Before this ODAC meeting, the FDA provided over 200 pages of pre-meeting materials, including the FDA’s data evaluation and the research materials provided by the pharmaceutical companies, which are highly valuable for analysis. Follow the WeChat official account “Medical Telescope” and reply “20240315” in the backend to obtain the complete briefing materials.
About the CARTITUDE-4 Study
A Phase III randomized, open-label trial (NCT04181827) enrolled lenalidomide-refractory MM patients and assigned them to receive Carvykti or physician’s choice of treatment. All patients had previously received 1-3 lines of therapy. The primary endpoint was PFS.
The study enrolled a total of 419 patients, randomized, with 208 receiving Carvykti and 211 receiving physician’s choice of treatment. Approximately 32% of patients had previously received one line of therapy. The median follow-up time was 15.9 monthsThe median PFS was not reached in the Carvykti group, while the median PFS in the physician’s choice group was 11.8 months (HR=0.26, 95% CI: 0.18-0.38, P<0.001). The OS results were not yet mature, and at the data cutoff date, HR=0.78 (95% CI: 0.5-1.2, P=0.26). The estimated 12-month OS rates were 84.1% and 83.6%, respectively.
About the KarMMa-3 Study
An international, open-label, Phase III clinical trial evaluated the efficacy and safety of ide-cel versus standard therapy in patients with R/R MM who had received 2-4 prior lines of treatment. The primary endpoint was PFS, and key secondary endpoints included ORR (PR or better) and OS.
A total of 386 patients were enrolled and assigned in a 2:1 ratio, with 254 receiving ide-cel treatment and 132 receiving standard treatment. The median follow-up time was 18.6 months. The median PFS was 13.3 months in the ide-cel group and 4.4 months in the standard treatment group (HR=0.49, 95% CI: 0.38-0.65, P<0.001) (see image below). 71% of patients in the ide-cel group achieved a response, compared to 42% in the standard treatment group (P<0.001). The CR rates were 39% and 5%, respectively. The OS data were not yet mature.
While these CAR-T therapies demonstrated long-term benefits, the experts at the ODAC meeting expressed concerns about the potential early risks and aimed to carefully evaluate the risk-benefit profiles before making their recommendations to the FDA. The agency will consider the advisory committee’s input, along with other available data, in making its final regulatory decisions on these product indications.
Reference Material
[1] FDA official website
[2] https://www.biospace.com/article/fda-adcomm-votes-on-bms-and-j-and-j-s-car-t-therapies-for-multiple-myeloma/
[3] https://www.fiercepharma.com/pharma/fda-flags-early-deaths-car-t-myeloma-trials-jj-and-legends-carvykti-bristol-myers-abecma
[4] Other public information
Content Cource:抗体圈